Figure 1
Structure de la peau La réparation cutanée Protéinases et remodelage tissulaire Le complexe Glycyl-L-Histidyl-L-Lysine-Cu2+
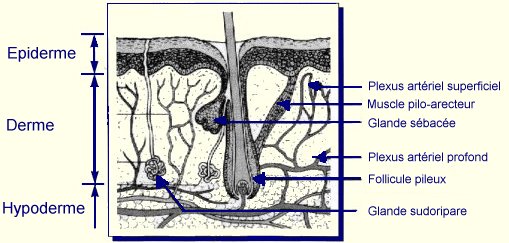
La peau, enveloppe extérieure du corps des animaux vertébrés, a pour fonction principale d’établir une barričre de protection contre les atteintes de l’environnement tout en permettant certains échanges entre le milieu intérieur et le milieu extérieur. Elle est le sičge de nombreux processus métaboliques qui sont modulés par les conditions physiologiques de l’organisme et les conditions de l’environnement. La peau est constituée de l’extérieur vers l’intérieur, de trois zones distinctes d’origines embryologiques différentes : l’épiderme (d’origine ectodermique), le derme et l’hypoderme (d’origine mésodermique).
L’épiderme constitue la couche la plus superficielle de la peau. C’est un épithélium pluristratifié, kératinisé, dont la face profonde est déprimée par des digitations (papilles dermiques). La face superficielle est plane et criblée de nombreux petits orifices correspondants aux ostiums pilaires et aux pores sudoraux qui sont des structures épithéliales spécialisées d’annexes épidermiques (respectivement les follicules pilo-sébacés et les glandes sudorales) logées en grande partie dans le derme et l’hypoderme (Grosshans, 1977).
Si différents types cellulaires coexistent dans l’épiderme, les kératinocytes sont largement majoritaires (90%). Ce sont des cellules épithéliales différenciées pour la synthčse des kératines qui représentent 95% des protéines totales de l’épiderme. Ces kératines sont des protéines fibreuses organisées en tonofilaments qui forment, avec les microfilaments d’actine et les microtubules, le cytosquelette des kératinocytes. La différenciation des kératinocytes s’accompagne d’un ensemble de transformations morphologiques et biochimiques qui aboutit ŕ la formation de cellules anucléées et aplaties qui desquament ŕ la surface de la peau, les cornéocytes. Ainsi, au cours de leur maturation cornée, les kératinocytes migrent de la profondeur vers la surface de l’épiderme (en environ 30 jours chez l’homme) et se répartissent en 4 couches
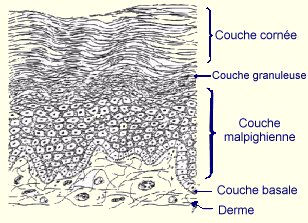
- la couche basale constituée d’une rangée unique de cellules cylindriques ou cubiques, qui jouent un rôle important dans la régénération épidermique.
- la couche malpighienne (ou stratum spinosum) constituée de 5 ŕ 10 couches cellulaires superposées qui s’aplatissent au fur et ŕ mesure de leur ascension.
- la couche granuleuse (ou stratum granulosum) formée de 1 ŕ 3 couches cellulaires disposées parallčlement ŕ la surface cutanée. Le caractčre essentiel des cellules de cette couche est la présence des grains de kératohyaline, principalement formés d’une protéine riche en histidine : la profilaggrine. Les derničres couches possčdent des kératinosomes qui interviennent dans la desquamation et la formation d’un manteau lipidique pericellulaire.
- la couche cornée (ou stratum corneum) formée de 5 ŕ 10 assises de cornéocytes, cellules lamelleuses aux limites cytoplasmiques indistinctes qui desquament ŕ la surface de la peau, assurant un rôle de barričre.
Ces différentes couches épidermiques sont intimement liées entre elles. La cohésion intercellulaire est renforcée par la présence de desmosomes dont le nombre augmente au cours de la différenciation. Le processus de kératinisation s’accompagne d’importantes modifications biochimiques telles que l’expression de nouvelles kératines acides et basiques et de nouvelles protéines de l’enveloppe des cornéocytes comme l’involucrine et la kératolinine qui sont des marqueurs de la différenciation terminale (Lepage, 1980).
D’autres types cellulaires s’organisent au sein de l’épiderme avec les kératinocytes (Kanitakis, 1995):
- les mélanocytes qui sont des cellules issues des crętes neurales et localisées dans la couche basale. Ces cellules sont responsables de la synthčse et de la maturation de la mélanine ŕ l’intérieur d’organites cellulaires particuliers : les mélanosomes. Ces mélanosomes sont transférés aux kératinocytes voisins oů ils forment une calotte "supranucléaire" protégeant le matériel génétique des effets mutagčnes des rayonnements ultraviolets.
- les cellules de Langerhans, d’origine monocytaire, qui sont localisées dans la partie profonde et moyenne de l’épiderme. Elles forment un réseau de cellules "sentinelles" chargées de présenter les antigčnes exogčnes déposés sur la peau aux lymphocytes, induisant ainsi une réponse immunitaire de type cellulaire.
- les cellules de Merckel, appartenant au systčme neuroendocrinien diffus, localisées dans la couche basale. Elles posséderaient un rôle de mécanorécepteurs, participeraient ŕ la formation du plexus nerveux superficiel et au positionnement de la partie superficielle des muscles pilo-érecteurs dans le derme.
- des fibres nerveuses, Leur présence, longtemps discutée au sein de l’épiderme, a été observée au contact des cellules de Langerhans dont elles pourraient moduler la fonction de présentation antigénique (Kanitakis, 1995).
L’épiderme possčde ou est parcouru par différentes annexes épidermiques comme les canaux excréteurs des glandes sudorales qui assurent un rôle fondamental dans la thermorégulation par le biais de la sudation ; les tiges des poils dont les follicules pileux sont localisés dans la partie profonde de l’hypoderme ; les ongles, tablettes dures recouvrant la derničre phalange des doigts et constitués de cornéocytes compacts.
La jonction dermo-épidermique appelée aussi membrane basale épidermique est une structure complexe séparant l’épiderme du derme (Briggaman, 1982, Stanley et coll., 1982b), élaborée ŕ la fois par les kératinocytes basaux et les fibroblastes dermiques. Elle peut ętre divisée en 4 zones allant de l’épiderme vers le derme (Fig 3) :
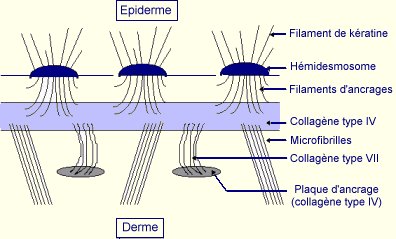
- la membrane plasmique des kératinocytes basaux avec leur structure d’attache : les hémidesmosomes. Ces derniers sont formés d'une plaque intracellulaire et de composants transmembranaire qui constituent un lien permettant l'attachement des kératinocytes basaux de l'épiderme au derme adjacent. Parmi les prinicpaux constituants des hémidesmosomes, différentes études ont permis de mettre en évidence la présence de deux antigčnes de la pemphigoďde bulleuse (BPAG-1 et BPAG-2). BPAG-1 est une protéine non collagénique qui constitue l'auto-antigčne majeur de la pemphigoďde bulleuse. BPAG-2 est une protéine collagénique transmembranaire, également nommée collagčne de type XVII (Li et coll., 1993, Li et coll., 1992). D'autres protéines sont également associées dans le complexe de l'hémidesmosome comme la plectine et l'intégrine a6b4 (Bernard, 1995). Celle-ci médie l'attachement des hémidesmosomes ŕ la membrane basale (Sonnenberg et coll., 1991).
- la lamina lucida traversée par des filaments d’ancrage (riche en laminine-1, -5 et - 6). Ces filaments, plus nombreux au niveau des hémidesmosomes, forment un complexe d'adhésion continu avec ces derniers en se liant ŕ la portion extracellulaire de l'intégrine a6b4 ŕ la surface des kératinocytes.
- la lamina densa, majoritairement constitutée de collagčne de type IV, forme la zone d’ancrage des filaments et des fibres issus de l’épiderme et de la zone fibrillaire. La lamina densa est également constituée de laminine-1, de nidogčne et de protéoglycannes.
- la zone fibrillaire qui contient des fibres d'ancrages reliant la lamina densa de la membrane basale ŕ des plaques d'ancrage dans le derme papillaire (Uitto, 1996). Les fibres d'ancrage sont constituées de collagčne de type VII (Burgeson, 1993).
Des études récentes montrent que l'organisation de la jonction dermo-épidermique pourrait impliquer des contacts entre les différents constituants beaucoup plus étroits que ce que l'on avait imaginé. Ainsi, le collagčne de type VII des fibres d'ancrage pourrait ętre en contact avec la laminine-5 des fibrilles d'ancrage (Chen et coll., 1999).
La jonction dermo-épidermique assure dans la peau des fonctions fondamentales : elle possčde un rôle de support mécanique pour l’adhésion de l’épiderme au derme et un rôle de barričre sélective permettant le contrôle des échanges moléculaires et cellulaires entre les deux compartiments. Elle est également, ŕ travers les glycoprotéines qui la constituent - en particulier les laminines - le support de l’adhésion et de la migration des kératinocytes lors de la restauration de l’intégrité épidermique (Zhang et Kramer, 1996), étape fondamentale de la cicatrisation cutanée.
Le derme, d’épaisseur trčs variable selon les régions du corps, est un tissu conjonctif fibro-élastique composé de cellules et de fibres baignant dans une substance amorphe appelée substance fondamentale. L’ensemble fibres et substance fondamentale est regroupé sous le nom de matrice extracellulaire.
Histologiquement, le derme est divisé en deux zones : une zone superficielle papillaire et une zone profonde réticulaire (Grosshans, 1977).
- Le derme papillaire est un tissu conjonctif lâche qui s’insinue entre les crętes de l’épiderme, formant ainsi les papilles dermiques. Il est composé d’un réseau de faisceaux de collagčne relativement fins et orientés perpendiculairement ŕ la membrane basale épidermique. Ces faisceaux baignent dans une substance interfibrillaire abondante constituée de protéoglycannes, de glycosaminoglycannes et de fibres élastiques. Cette partie du derme est riche en vaisseaux sanguins et en cellules, principalement des fibroblastes.
- Le derme réticulaire profond se différencie du derme papillaire par un tissu conjonctif dense constitué de faisceaux de fibres de collagčnes entremęlées ŕ des fibres élastiques qui suivent généralement l’orientation des faisceaux de collagčnes, de plus en plus épais vers la profondeur du derme. Le derme réticulaire contient peu de substance fondamentale et peu de cellules conjonctives.
La matrice extracellulaire du tissu conjonctif dermique est constituée de quatre types de macromolécules : les collagčnes, l’élastine, les glycoprotéines de structure et les protéoglycannes. La nature et la quantité de ces composants régissent les propriétés mécaniques de la peau normale et sont ŕ l’origine des modifications physiopathologiques les plus visibles du relief cutané.
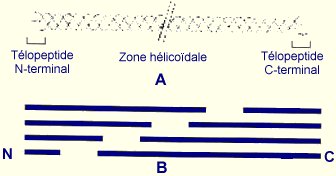
Les collagčnes représentent les protéines les plus abondantes du derme (70 ŕ 80%) (Van der Rest, 1993). Ce terme regroupe une famille de glycoprotéines qui ont toutes en commun leur présence dans les tissus conjonctifs et au moins un domaine caractéristique appelée triple hélice polypeptidique ("domaine collagénique"). Cette triple hélice est constituée de trois chaînes polypeptidiques (appelées chaînes a) identiques ou différentes, enroulées en hélice sur elles-męmes et les unes autour des autres. Cette triple hélice peut comporter, suivant le type de collagčne, un nombre variable d’interruptions non hélicoďdales et des extrémités globulaires plus ou moins volumineuses ("domaines non collagéniques"). Cet arrangement spatial particulier est rendu possible par la présence dans la structure primaire des chaînes a d’un résidu glycine tous les trois acides aminés [(Gly-X-Y)n]. Dans ces triplets, X et Y sont fréquemment des résidus de proline et d’hydroxyproline respectivement. Cette organisation en triplets confčre sa rigidité ŕ la molécule de collagčne, contribuant ainsi ŕ la fonction mécanique de celle-ci. La conformation en triple hélice aboutit nécessairement ŕ une protéine fibrillaire capable de se polymériser en fibres, en filaments ou lames (Figure 4). Cette particularité permet d'exclure des protéines apparentées aux molécules de collagčnes telles que le constituant C1q du complément et l'enzyme acétyl-cholinestérase de la plaque motrice (Borel, 1991).
|
Figure 4
|
||
| A : Monomére de collagène de type I | B : Réseau formé par l'association covalente des molécules de collagène de type I |
|
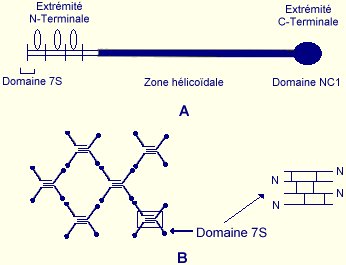
Dans le cas du collagčne de type IV, la structure particuličre du monomčre lui permet de s’organiser en un réseau plan permettant la formation des membranes basales (Figure 5).
|
Figure 5
|
||
| A : Monomére de collagène de type IV | B : Réseau plan formé par l'association covalente des molécules de collagène de type IV |
|
Actuellement, on dénombre 19 types de collagčne dont la moitié au moins est présente dans la peau. Ces molécules sont génétiquement distinctes et diffčrent aussi par leur mode de synthčse, leur répartition tissulaire, leur taille, leur forme et leurs fonctions biologiques.
Le tableau I résume la nomenclature, la composition des chaînes et leur distribution tissulaire. Les collagčnes dont la localisation dermique est reconnue regroupent :
- des collagčnes fibrillaires (type I,III,V) qui représentent au moins 80 ŕ 90% du collagčne dermique total. Ils constituent le composant essentiel du réseau fibreux et possčdent un rôle mécanique. Il est ŕ noter que les proportions relatives de collagčnes de type I et III sont un facteur déterminant de la fonction tissulaire. Une diminution du rapport type III/type I se traduit par une perte des propriétés biomécaniques de la peau et notamment une diminution de sa flexibilité et une augmentation de sa fragilité, ainsi que celle des vaisseaux sanguins.
- un collagčne de membrane basale (type IV) trouvé au niveau des plaques d’ancrage de la membrane basale épidermique ainsi que dans les membranes basales entourant les annexes épidermiques et les structures vasculaires.
- des collagčnes FACIT (Fibril-Associated Collagens with Interrupted Triple Helices) dont le rôle est d’associer les fibres ŕ leur environnement (collagčnes de type VIII, XII, XIV, XV et XVI),
- des collagčnes de type VI et VII, dont les structures et les propriétés les rendent difficiles ŕ classer dans une des ces catégories précédentes. Le collagčne de type VI constitue un fin réseau fibreux qui réunit et fixe les éléments de structure du derme et de l’hypoderme (cellules, grosses fibres, vaisseaux sanguins, filet nerveux). Le collagčne de type VII constitue l’essentiel des fibrilles d’ancrage permettant l’accrochage de la membrane basale au derme sous-jacent.
L’élastine est le constituant majeur (90%) des fibres élastiques du derme adulte. Elle est toujours associée ŕ des structures microfibrillaires. L’ensemble représente 2 ŕ 4 % du volume du derme et confčre ŕ la peau ses propriétés d’élasticité et de souplesse.
L’élastine est synthétisée sous forme d’un monomčre de 72 kDa appelé tropoélastine. Lors de la maturation de l’ARN préméssager codant ce monomčre, certains exons peuvent ętre excisés, conduisant ainsi ŕ la formation d’isoformes ayant des propriétés chimiques, physiques et fonctionnelles distinctes.
Les fibres élastiques sont formées par des monomčres de tropoélastine unis par des liaisons hydrophobes et dont l’ensemble est stabilisé par des liaisons croisées covalentes (desmosine et isodesmosine). En outre, des structures microfibrillaires constituées de glycoprotéines entourent les agrégats d'élastine, ancrant les fibres élastiques dans le derme papillaire. Ces structures microfibrillaires sont constituées d'association de glycoprotéines telles les fibrillines (Sakai et coll., 1991) et les MAGP (Microfibril Associated Glycoprotein) (Cleary, 1996). Les fibres élastiques du derme réticulaire sont plus épaisses et plus anastomotiques que celles du derme superficiel.
Dans les années 70, de trčs nombreuses glycoprotéines ont été isolées ŕ partir de la matrice extracellulaire. L'une des caractéristiques communes de ces glycoprotéines est d'ętre composées par des motifs structuraux répétés formant ensemble des domaines qui confčrent ŕ ces molécules leurs différentes fonctions (Johansson 1996). Le tableau II présente les principales glycoprotéines de la matrice extracellulaire (Johansson, 1996, Raghow, 1994, Yamada et Clark, 1996).
Ces molécules jouent un rôle dans les interactions cellules-matrice, en particulier dans les phénomčnes d’adhésion et de migration cellulaire. Il est ici important de noter que la fibronectine est une composante essentielle de la matrice provisoire qui est mise en place lors des phénomčnes de réparation tissulaire (O'Keefe et coll., 1985, Gailit et coll., 1993).
Les protéoglycannes sont des macromolécules complexes de grande taille dont l’hydratation assure en grande partie la tonicité de la peau. Ils sont dispersés entre les fibres du tissu conjonctif dermique.
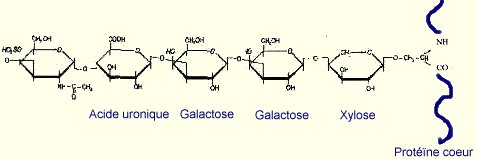
Les protéoglycannes sont formés d’une chaîne polypeptidique appelée protéine cśur sur laquelle sont greffés, par l’intermédiaire de liaisons covalentes, des polymčres formés de résidus osidiques souvent aminés ou sulfatés appelés glycosaminoglycannes (GAGs). Les GAGs que l’on retrouve dans le derme sont le chondroďtine-6-sulfate (CS), le dermatanne-sulfate (DS) et l’héparanne-sulfate (HS) (Figure 6). La liaison des chaînes osidiques se fait sur un résidu sérine de la protéine cśur. Dans la cas le plus fréquent, un tétrose constitué de deux galactoses, d’un xylose et d’un acide uronique sert ŕ relier les chaînes glycaniques ŕ la sérine (Figure 7) (Borel, 1997). On trouve également un GAG non sulfaté de trčs haute masse moléculaire, l’acide hyaluronique (HA), qui semble promouvoir la migration cellulaire, par l’intermédiaire de récepteurs membranaires tels le CD44 et les RHAMM (Receptor for Hyaluronan Mediated Motility) situés ŕ la surface des fibroblastes (O'Keefe et coll., 1985). Les propriétés biologiques et physico-chimiques des protéoglycannes sont le reflet de leur composition en GAGs, fortement sulfatés ou non, et de la nature de leur protéine porteuse. Ainsi, les trčs nombreuses fonctions acides qu’ils portent confčrent aux protéoglycannes des charges négatives permettant de "piéger" les ions, l’eau et divers métabolites, contribuant ainsi ŕ l’hydratation des tissus et ŕ la résistance aux forces de pression et d’étirement.
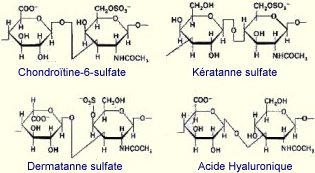
Figure 6
Le tableau III présente les principaux protéoglycannes clonés chez les mammifčres (Herbage, 1995, Iozzo, 1997).
Certains des protéoglycannes sont capables de s'agréger entre eux et/ou avec d’autres molécules de la matrice extracellulaire (collagčnes, laminines, fibronectine), pouvant ainsi intervenir dans les interactions cellule-matrice. Ils peuvent également se lier ŕ des facteurs de croissance ou se comporter en co-récepteurs de ces derniers. En étant ainsi capables d’interagir avec les facteurs de croissance, les protéoglycannes sont capables d’intervenir dans la modulation des comportements cellulaires, comme la prolifération, la différenciation, la migration,...(Gallo et coll., 1996)
La population largement majoritaire du derme est constituée de fibroblastes. Ce sont de grandes cellules (100 µm) fusiformes ou étoilées, possédant de long prolongements cytoplasmiques. Elle renferment un noyau unique médian et ovale, contenant un ou plusieurs nucléoles. Pauvre en organites cytoplasmiques au repos, le fibroblaste en activité possčde ŕ l’inverse de nombreux organites cellulaires. Ainsi, l’observation en microscopie électronique permet de distinguer un réticulum endoplasmique granuleux abondant, un appareil de Golgi développé, des ribosomes libres ou en amas nombreux. L’ensemble témoigne d’une grande activité de synthčse des fibroblastes. Ces derniers produisent et sécrčtent en effet la majorité des molécules constituant la matrice extracellulaire notamment les collagčnes, l’élastine, les glycoprotéines de structure et les protéoglycannes mais aussi des protéinases capables de dégrader et de remodeler cette matrice, telles les métalloprotéinases matricielles (MMPs) que nous détaillerons plus loin.
Le derme contient également des dendrocytes dermiques, qui représentent une population hétérogčne de cellules mésenchymateuses dendritiques précédemment confondues avec des fibroblastes. Elles sont trouvées dans le derme papillaire en position périvasculaire et dans le derme profond autour des annexes épidermiques.
A côté de ces cellules spécifiques, on trouve des cellules d’origine sanguine telles que les mastocytes, cellules intervenant dans les réactions allergiques, retrouvées en petit nombre autour des capillaires dermiques ; les macrophages qui possčdent un rôle crucial dans l’inflammation, la détersion du tissu et l’activation de l’immunité.
Le derme contient également des cellules endothéliales qui participent ŕ la formation des vaisseaux et des cellules nerveuses assurant les fonctions sensorielles du tissu cutané.
Le systčme vasculaire présent au niveau du derme intervient dans la thermorégulation, la régulation de la pression artérielle et la défense immunitaire de l’organisme. Il est formé d’artčres sous-cutanées qui cheminent dans les fascias et envoient des colatérales qui traversent l’hypoderme et atteignent le derme profond oů elles s’anastomosent pour former le plexus artériel profond, disposé horizontalement : celui-ci envoie des artčres "en candélabre" qui remontent verticalement dans le derme et forment dans la zone sous papillaire le plexus artériel superficiel. De ce plexus, se détachent des artérioles précapillaires qui donnent naissance aux anses capillaires comportant un bras ascendant et un bras descendant (veinule postcapillaire). Le réseau veineux comporte également un plexus veineux sous papillaire et un plexus veineux profond.
L’innervation cutanée est riche et complexe. La voie afférente, constituée d’un nerf sensitif du systčme cérébro-spinal, est impliquée dans la perception des variations et agressions extérieures comme le toucher, les vibrations, la pression, la température, la douleur,... Cette fonction est assurée par un réseau composé de fibres myélinisées, de terminaisons nerveuses libres et de corpuscules tactiles (corpuscules de Wagner-Meissner et de Vater-Pacini, localisés dans les papilles dermiques et la jonction dermo-épidermique respectivement). La voie efférente comporte des fibres amyéliniques du systčme sympathique qui régulent la vasomotricité, la sécrétion sudorale et la pilo-arrection (Kanitakis, 1995).
L’hypoderme est un tissu adipeux séparant le derme et les plans aponévrotiques et musculaires sous-jacents. Il est constitué de septa conjonctifs qui cloisonnent les lobes adipeux. Les cellules adipeuses qui le constituent sont des cellules mésenchymateuses différenciées, rondes ou polygonales, possédant un noyau triangulaire périphérique et un cytoplasme riche en triglycérides et en acides gras.
Outre son rôle énergétique dű ŕ la reserve de graisse qui le constitue, l'hypoderme possčde également un rôle de protection mécanique et thermique.
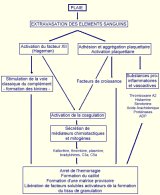
Toute lésion tissulaire, qu’elle soit d’origine mécanique, chimique, thermique ou immunologique provoque une cascade de réponses qui conduit ŕ la formation d’un tissu de réparation et ŕ la cicatrisation de la blessure. Cette cicatrisation nécessite l’intégration de nombreux processus dynamiques impliquant ŕ la fois la matrice extracellulaire du tissu lésé, des médiateurs solubles ainsi que des cellules sanguines et mésenchymateuses. Les processus de réparation tissulaire suivent une cinétique spécifique qui permet de distinguer trois phases (Clark, 1985):
La séparation chronologique de ces trois phases reste toutefois trčs théorique, puisque dans la réalité des processus cicatriciels, elles se chevauchent partiellement voire largement.
Premičre phase de la réparation tissulaire, la phase inflammatoire
limite les perturbations physiologiques provoquées par la lésion en stoppant
l’hémorragie, nettoie le foyer de blessure, fournit une matrice provisoire pour
la migration des cellules et génčre des facteurs qui participeront ŕ l’initiation
de la formation du tissu de granulation.(Figure 8)
Toute blessure provoque la rupture de parois vasculaires et donc l’extravasation d’éléments sanguins. Ce contact entre le sang et les tissus sous-jacents est l’événement critique initial de la cicatrisation qui provoque l’activation d’un grand nombre de systčmes tels que l'agrégation plaquettaire et la cascade de la coagulation qui aboutissent ŕ la formation du caillot sanguin .
Ainsi, les plaquettes sanguines, stimulées par des composants générés ou exprimés sur le site de la lésion, tels les fragments de collagčnes fibrillaires ou la thrombine, adhérent au foyer de la blessure et s'agrčgent, constituant le "clou plaquettaire" sur lequel le caillot sanguin vient se former (Borel, 1998).
La fuite des protéines et des éléments figurés du sang ŕ partir des vaisseaux sanguins lésés conduit ŕ la coagulation par différentes voies (Figure 9)(Majno, 1996) :
Au terme d’une cascade d’activation, le fibrinogčne est clivé pour donner des monomčres de fibrine qui polymérisent pour former un réseau dense autour des hématies, aboutissant ainsi ŕ la formation d’un caillot obturant mécaniquement les brčches faites dans les vaisseaux sanguins. L’ensemble aboutit ŕ la formation d’un caillot constitué ŕ 94,9% de fibrine, ŕ 4,4% de fibronectine et ŕ 0,7% d’inhibiteurs de la plasmine .
La coagulation s'arręte lorsque les stimuli initiateurs disparaissent. De plus, les activités intrinsčques des vaisseaux sanguins limitent l'agrégation plaquettaire et la coagulation ŕ la zone lésée. Ces activités incluent la production de NO et de prostaglandine PGI2, tous deux inhibiteurs de l'agrégation plaquettaire, l’inactivation de la thrombine par l’antithrombine III, l'activation de la protéine C, une enzyme capable de dégrader les facteurs Va et VIIIa de la coagulation, la libération d’activateurs du plasminogčne qui initient la lyse du caillot en convertissant le plasminogčne en plasmine (Moatti, 1998).
Outre son rôle dans l’établissement du caillot de fibrine, la coagulation participe ŕ la réponse inflammatoire par différentes voies. Ainsi, l’activation du facteur XII permet la libération de bradykinine, puissant vasodilatateur, et l’activation de la cascade du complément (également activé par contact). Certains des facteurs du complément ainsi générés, comme le C3a et le C5a, augmentent directement la perméabilité vasculaire, recrutent les neutrophiles et les monocytes au niveau du foyer de blessure, stimulent la libération de médiateurs vasoactifs (histamine, leucotrienne C4 et D4) par les mastocytes et de radicaux libres oxygénés par les neutrophiles et les macrophages. Les différents rôles de la coagulation sont présentés dans le Tableau IV (Clark, 1995).
On peut en particulier noter que le caillot fournit un réseau matriciel facilitant le recrutement et la migration des cellules vers le site lésé. Ainsi, la fibrine et la fibronectine agissent comme une matrice provisoire permettant l’influx de neutrophiles, de fibroblastes, et de cellules épithéliales (Brown et coll., 1993b). Ces cellules interagissent avec le caillot via des récepteurs de type intégrine capables de lier la fibrine, la fibronectine et la vitronectine (Ginsberg et coll., 1992).
Différents types cellulaires sont impliqués dans la phase inflammatoire de la réparation tissulaire. Certaines de ces cellules sont résidentes au niveau du foyer de blessure tandis que d’autres sont recrutées ŕ partir du compartiment vasculaire ou des tissus voisins.
L’activation des plaquettes provoque la libération de nombreuses substances favorisant l’inflammation (Borel, 1998) :
Les mastocytes sont les premičres cellules qui semblent intervenir aprčs les plaquettes. Leur fonction paraît d’autant plus importante que ce sont des cellules résidentes du derme, attirées dans le foyer de blessure et activées, ipso facto, sécrétant des facteurs stimulants pour d’autres cellules et susceptibles de participer ŕ la réparation. Ils agissent ŕ tous les stades, jusqu'ŕ complčte restitution du tissu lésé, ŕ la différence des autres types cellulaires qui exercent leur fonction pendant un intervalle de temps limité.
Les mastocytes sécrčtent des enzymes protéolytiques, telles la tryptase et la chymase qui permettent l’activation d’autres protéinases impliquées dans la détersion de la plaie et qui libčrent ŕ partir des protéines tissulaires, des peptides doués d’activité chimiotactique, attirant ainsi tous les autres types cellulaires (Levi-Schaffer, 1995). Ces cellules sécrčtent également des facteurs activant la migration des fibroblastes vers le tissu lésé et stimulant la production de collagčnes. Parmi ces cytokines, l’interleukine-4 (IL-4) semble occuper une place prépondérante (Gillery et coll., 1992a).
Les polynucléaires neutrophiles sont les premičres cellules ŕ infiltrer le site de la blessure. Ils sont attirés sur ce site par les produits de l’activation plaquettaire (kallicréine, fibrinopeptides, produits de dégradation de la fibrine, C5a, leucotrienne B4, PDGF, PF-4), par les peptides de dégradation des bactéries (formyl-peptides), ou par des facteurs libérés par les cellules endothéliales (Platelet-Activating Factor : PAF). En plus de stimuler leur migration des polynucléaires, ces peptides augmentent l’expression des récepteurs CD11/CD18 médiant l’adhésion des neutrophiles aux cellules endothéliales vasculaires, facilitant ainsi leur transmigration ŕ travers l’endothélium vasculaire. Cette transmigration est également favorisée par une libération accrue d’élastase et de protéinases par les polynucléaires neutrophiles activés (Stahle-Backdahl et coll., 1994, Stahle-Backdahl et Parks 1993).
L’activation des neutrophiles induit la phagocytose et l’élimination des débris cellulaires et tissulaires. Ils contribuent ŕ la détersion des tissus nécrosés, grâce ŕ leur sécrétion de protéinases, d’oxydases et de radicaux libres oxygénés. Toutefois, l’introduction d’antisérum antipolynucléaires neutrophiles dans une plaie expérimentale aseptique chez le cobaye ne retarde pas la cicatrisation (Simpson et Ross, 1972).
Deux jours aprčs la survenue de la blessure, la fin de l’infiltration des neutrophiles, leur entrée en apoptose et leur phagocytose par les macrophages tissulaires marque la fin de la phase inflammatoire précoce de la cicatrisation.
Les macrophages suivent immédiatement l’invasion tissulaire par les polynucléaires. Ils sont attirés au site de blessure par différentes classes de chimioattractants : des dérivés de la matrice extracellulaire du tissu conjonctif (fragments de collagčne, d’élastine, de fibronectine), des dérivés des bactéries, des protéines diverses (C5a, C5a desArg, Fibrinopeptides, thrombine) ou encore des chimiokines (LB4, MCP, RANTES, MIP 1a et 1b) ou facteur de croissance (PDGF, TGF-b). Comme pour les neutrophiles, les chimioattractants stimulent l’adhésion des monocytes ŕ l’endothélium des vaisseaux sanguins et leur migration ŕ travers la paroi vasculaire (Clark, 1985). Toutefois, il existe également une faible proportion de macrophages résidents (histiocytes), présents normalement dans le tissu conjonctif.
La liaison des monocytes ou des macrophages aux protéines de la matrice extracellulaire par des récepteurs de type intégrine stimule la phagocytose des débris tissulaires ainsi que des éventuels micro-organismes étrangers infectant la plaie. Le premier rôle des macrophages est donc clairement la détersion du foyer de blessure. Cette fonction peut ętre facilitée par la capacité des macrophages ŕ libérer des enzymes de dégradation telles que les métalloprotéinases matricielles (MMP-1,-3, -9) (Welgus et coll., 1990) ou l’élastase (Shapiro et coll., 1993, Shapiro et coll., 1992) ainsi que des intermédiaires oxygénés et des éicosanoďdes .
Les macrophages produisent aussi de nombreux médiateurs solubles capables de moduler la réponse inflammatoire, soit en l’amplifiant (Thromboxanes A2 : TXA2, prostaglandines, leukotrienes, Tumor Nécrosis Factor-a: TNF-a, IL-1 a et b, IL-6), soit en l’inhibant (PGI2). Le Granulocyte/Macrophage-Colony Stimulating Factor (GM-CSF), également produit, induit la différenciation des cellules souches en granulocytes et en macrophages.
Ces macrophages sont également capables de sécréter un grand nombre de chimiokines, cytokines, facteurs de croissance (Martin, 1997). Ces facteurs apparaissent nécessaires pour l’initiation et la formation d’un nouveau tissu. En effet, la déplétion en macrophages d’une plaie in vivo, ŕ l'aide d'un antisérum spécifique, provoque un retard dans la formation et l’extension du tissu de granulation (Leibovich et Ross 1975).
Les lymphocytes T apparaissent plus tardivement dans la plaie, ils ne sont pas essentiels ŕ l’initiation de la cicatrisation, qui peut se faire en leur absence, mais sont nécessaires ŕ l’obtention d’une réparation normale (Barbul et coll., 1989). Ils atteignent un maximum vers le sixičme jour. In vivo, la déplétion en lymphocytes T par des anticorps spécifiques provoque une diminution de l'accumulation de collagčnes et une détérioration de la qualité de la cicatrisation mesurée par l’étude de la résistance cutanée (Peterson et coll., 1987). Si la diminution des lymphocytes T-helper ne provoque pas de modification des processus de cicatrisation, celles des T-supresseurs se traduit par une augmentation de l'accumulation de collagčnes et des forces de tension cutanée (Barbul et coll., 1989).
Les lymphocytes T sont capables sous l’action de facteurs solubles ou d’antigčnes présents dans la plaie, de recruter et d’activer les fibroblastes responsables de la synthčse de collagčnes. Ils peuvent agir indirectement sur ces derniers en stimulant les macrophages, ou directement par l’intermédiaire de diverses cytokines telles le Lymphocyte Derived Chemotactic Factor for Fibroblasts (LDCD-F), le Fibroblast Activating Factor (FAF), le TGF-b.
Aprčs détersion de la plaie, la stimulation antigénique diminue et les T-helper sont remplacés par les T-supresseurs qui exercent un contrôle négatif. La balance T-helper/T-supresseurs influence l’activité cellulaire cicatricielle et une rupture de cet équilibre pourrait ętre responsable de la fibrose (Wahl, 1988).
L’ensemble de ces phénomčnes concourent ŕ la mise en place d’une matrice provisoire dans laquelle baignent les cellules qui libčrent de nombreux médiateurs solubles comme les facteurs de croissance intervenant dans les étapes ultérieures de la cicatrisation.
La réépithélialisation peut ętre définie comme la reconstitution d’un épithélium pluristratifié, recouvrant la plaie et ayant retrouvé sa fonction de barričre, son caractčre pigmenté mais aussi ses fonctions sensorielles et immunitaires. Elle implique donc des phénomčnes de migration et de prolifération cellulaire - en particulier celle des kératinocytes - de stratification et de maturation de l’épiderme néoformé, ainsi que la reformation d’une membrane basale complčte et la repopulation par les cellules spécialisées de l’épiderme (cellules de Merckel, de Langerhans et mélanocytes).
La migration cellulaire est un événement précoce et indispensable ŕ la réépithélialisation. Les événements initiant cette migration sont encore mal connus ; cependant la perte de contact avec les cellules voisines, due ŕ la blessure, est souvent évoquée ("free-edge effect"). Une faible concentration en Ca2+ dans le milieu semble également favoriser cette migration (Hennings et coll., 1980). Plus récemment, les conditions d’hypoxies dans lesquelles se retrouvent les cellules épithéliales aprčs la survenue d’une blessure ont également été évoquées comme étant l’un des éléments capable de participer ŕ l’initiation de la migration (Woodley, 1996), ainsi que la production par les fibroblastes du tissu conjonctif sous-jacent de divers facteurs de croissance et cytokines (voir plus loin).
Dčs la 6čme heure aprčs l’induction de la blessure, les kératinocytes des berges de la plaie commencent ŕ migrer latéralement le long de la matrice provisoire constituée essentiellement de fibrine et de fibronectine.
Le passage de l’état stationnaire ŕ l’état migrant s’accompagne de profondes modifications dans le phénotype du kératinocyte. Ainsi, de cellules polarisées, juxtaposées sur la membrane basale épidermique, ils deviennent plats et allongés. Les structures d’attachements membranaires responsables des liaisons cellule-cellule et cellule-matrice extracellulaire (desmosomes et hémi-desmosomes) disparaissent, les tonofilaments se désorganisent (Odland et Ross, 1968). Ces modifications provoquent une perte de la polarité cellulaire et la formation de lamellipodes permise par une augmentation de l’expression d’actine et d’a-actinine. Il est aussi ŕ noter que les cellules épithéliales migrantes situées en avant de la migration présentent un phénotype hautement phagocytaire. Toutefois, la phagocytose ne semble pas stimuler directement la migration (Takashima et Grinnell, 1984).
La migration des kératinocytes implique l’adhésion de ces cellules sur leur substratum. Il est aujourd’hui clairement établi que les kératinocytes sont capables d’adhérer et de migrer sur différentes molécules de la matrice extracellulaire. C’est par exemple le cas sur la fibronectine qui constitue la premičre matrice provisoire de la blessure. Cette liaison fait intervenir la séquence tripeptidique RGD de la fibronectine - RGD étant un motif de reconnaissance pour les récepteurs de type intégrine. Différentes expériences d’inhibition de liaison par compétition de séquence ou par anticorps spécifiques ont permis d’identifier l’intégrine a5b1 comme étant le récepteur de la fibronectine ŕ la surface des kératinocytes migrants (Kim et coll., 1992a, Kim et coll., 1992b).
Lors de leur migration, les kératinocytes sont également susceptibles de se trouver au contact de la vitronectine. Les męmes expériences que celles décrites précédemment ont permis de montrer que le récepteur en cause était l’intégrine de type avb5 (Kim et coll., 1994). En outre, il a également été montré que la présence de vitronectine dans la matrice était indispensable ŕ l’étalement des kératinocytes sur la fibronectine ou le collagčne (Stenn, 1987).
Durant leur migration, les kératinocytes sont également en contact avec le collagčne de membranes basales (type IV) et des collagčnes fibrillaires (type I et III). A l’inverse de la mobilité sur la fibronectine et bien que ces collagčnes en possčdent, cette migration ne fait pas intervenir le triplet RGD (Kim et coll., 1992b). Le récepteur des collagčnes fibrillaires sur les kératinocytes a été identifié comme étant l’isoforme a2b1 des intégrines (Chen et coll., 1993).
Le comportement des kératinocytes vis ŕ vis des laminines, glycoprotéines avec lesquelles ils sont en contact au niveau de la lamina lucida de la membrane basale, semble différent. En effet, les laminines inhibent de façon dose dépendante la migration des kératinocytes sur le collagčne (Woodley, 1988, Woodley, 1996). En fait, les laminines et, en particulier la laminine-5, seraient le facteur d’adhésion majeur des kératinocytes via l’intégrine a3b1 (Kim et coll., 1992b). Ainsi, des auteurs ont pu observer in vivo que la laminine n’apparaît pas au niveau de la jonction dermo-épidermique tant que la migration de l’épithélium n’est pas complčte (Clark et coll., 1982). Toutefois, ces résultats restent encore controversés (Zhang et Kramer, 1996.)
Les processus de migration des kératinocytes sont également rendus possible par la libération de métalloprotéinases matricielles au niveau du site lésé. Les kératinocytes eux-męmes expriment et sécrčtent lors de la migration de la MMP-1 (Sudbeck et coll., 1997, Saarialho-Kere et coll., 1995, Mignatti, 1996). Il semble que le degré de synthčse de MMPs soit directement relié ŕ la migration (Petersen et coll., 1990).
La prolifération des kératinocytes qui se produit aprčs 48 ŕ 72 heures, ne semble pas affecter la migration. Elle s’effectue sous l’influence de nombreux facteurs qui peuvent ętre sécrétés par les cellules avoisinantes ou par les kératinocytes eux-męmes : Fibroblast growth Factor : FGF-7 (Kératinocyte growth Factor : KGF), Epidermal Growth Factor (EGF), IL-1, IL-6, IL-8, Colony Stimulating Factor (CSF), TGF a et b , PDGF, TNF-a, Insulin-like Growth Factor : IGF-1. L’influence de ces facteurs a pu ętre vérifiée in vivo dans différents modčles animaux (Mustoe et coll., 1987, Brown et coll., 1989).
Parmi ces facteurs, certains sont également capables d’influencer la migration. C’est le cas pour l’EGF et le TGF-a qui la stimulent en augmentant l’expression de l’intégrine a2b1 ŕ la surface des kératinocytes et pour l’IL-1 par un mécanisme indépendant des intégrines (Chen et coll., 1995) qui semble davantage impliquer une surexpression de protéinases. D’autre part, il a été démontré que le TGF-b inhibait fortement la prolifération des kératinocytes sans en affecter la migration.
Les cellules épidermiques migrantes ne sont pas totalement différenciées, comme le sont les kératinocytes des couches supérieures de l’épiderme normal. Par exemple, les cellules migrantes ne contiennent pas les kératines ni la filagrine normalement exprimées dans un épiderme stratifié mature. En revanche, toutes les cellules migrantes contiennent les kératines spécifiques des cellules basales.
La maturation se fait simultanément avec la fermeture de la plaie et correspond ŕ une reprise de la fonction et de la morphologie normale des kératinocytes. Ainsi, la réapparition des kératosomes signe la restauration de l’imperméabilité ŕ l’eau ; la filagrine elle, réapparaît lorsque la migration est complčte (Lacour et Ortonne, 1983) : elle est d’abord détectée sur les bords de la cicatrice et correspond au retour des grains de kératohyaline.
L’utilisation de modčles animaux a permis de montrer que l’expression des différentes molécules composant la membrane basale épidermique étaient exprimées selon une séquence ordonnée (Woodley, 1996). Une étude réalisée sur la peau humaine a pu préciser que l’antigčne de la pemphigoďde bulleuse (BPAG-1) apparaissait en premier suivi par le collagčne de type IV, la laminine et le collagčne de type VII (Cornelius, 1986). La synthčse de ces différentes macromolécules, qui peut ętre effectuée par les fibroblastes ou par les kératinocytes (Stanley et coll., 1982a), est sous le contrôle de nombreux facteurs de croissance et cytokines tels le TGF-b, le TNF-a et l’IL-1. Le dépôt commence par les bords de la plaie et progresse vers le centre fixant l’épiderme au derme sous-jacent pendant que la fibrine et la fibronectine disparaissent progressivement. Lorsque la réépithélialisation est complčte, les cellules retrouvent leur phénotype normal et forment des hémidesmosomes qui les relient aux membranes basales. Toutefois, ces processus sont tardifs, l’adhérence derme-épiderme reste faible durant une longue période allant de plusieurs mois ŕ plusieurs années aprčs la réépithélialisation apparente complčte de la blessure (Woodley et coll., 1990).
Le tissu de granulation qui apparaît environ quatre jours aprčs la survenue de la blessure, contient des cellules, de la matrice extracellulaire et des vaisseaux sanguins. Sa formation repose sur les macrophages, le recrutement et la prolifération de fibroblastes qui vont produire un tissu conjonctif lâche, et sur la formation de nouveaux vaisseaux sanguins (angiogénčse) qui apportent l’oxygčne et les nutriments permettant de soutenir le métabolisme cellulaire. Ces composantes sont capables d’interagir entre elles : ainsi, les macrophages agissent sur les fibroblastes en synthétisant et en sécrétant de façon continue des cytokines et des facteurs de croissance capables de moduler leur comportement. Les fibroblastes, s’ils produisent, déposent et remodčlent la matrice extracellulaire, sont influencés par cette męme matrice qui régule les fonctions précédemment citées (Gillery et coll., 1996, Grinnell, 1994). Ces interactions entre la matrice extracellulaire et les fibroblastes évoluent pendant tout le développement du tissu de granulation : on parle de réciprocité dynamique.
La fibroplasie désigne l'existence d'un mélange de fibroblastes et de matrice extracellulaire. Sa formation est permise et contrôlée par de nombreuses cytokines qui peuvent ętre synthétisées et/ou libérées par les plaquettes et les macrophages dčs les premiers jours de la cicatrisation. Toutefois, les fibroblastes peuvent aussi produire certaines de ces cytokines auxquelles ils répondent de façon autocrine. Les principales cytokines et facteurs de croissance retrouvés au niveau du foyer de blessure et impliqués dans la formation du tissu de granulation sont regroupés dans le Tableau V (Borel, 1998, Martin, 1997) ; leur différents rôles sont illustrés figure 10. Pour simplifier, nous dirons que ces cytokines agissent en synergie pour permettre la migration des fibroblastes vers le site lésé, leur prolifération, la synthčse des différents composants de la matrice extracellulaire et l’angiogénčse.
La migration des fibroblastes résulte de l’interaction avec la matrice extracellulaire. Cette derničre constitue le support de cette migration via les différents récepteurs de type intégrine des fibroblastes, en particulier a4b1 pour la migration sur la fibronectine (Gailit et coll., 1993) et a1b1 ou a2b1 pour la migration sur le collagčne (Ignatius et coll., 1990, Staatz et coll., 1989). Elle fournit également une véritable information de direction aux cellules. Les fibroblastes ont en effet tendance ŕ s’aligner et ŕ migrer ŕ l’intérieur des discontinuités des molécules sur lesquelles elles ont adhérées. Cultivées in vitro sur de la fibronectine, les fibroblastes migrent ainsi le long et non au travers des fibres (Hsieh et Chen, 1983).
Cette migration implique également un systčme actif de protéolyse. De nombreuses enzymes produites par les fibroblastes, en coordination avec la plasmine provenant du sang, semblent ainsi impliquées (activateurs du plasminogčne, MMP-1, -2, -3). Certains facteurs chimiotactiques, tels le PDGF et le TGF-b, pourraient agir en stimulant la production et la sécrétion de ces protéinases (Laiho et coll., 1986, Overall et coll., 1989b).
Dans les différents événements impliqués dans la formation du tissu de granulation, l’un des plus cruciaux semble ętre la différentiation phénotypique d’une certaine fraction de la population des fibroblastes en myofibroblastes (Gabbiani et coll., 1971). La différence essentielle entre ces deux types cellulaires est la présence dans les myofibroblastes de faisceaux de microfilaments d’actine de type musculaire lisse (a-sm-actine),. Ils possčdent de nombreuses connexions entre eux et avec certains composants de la matrice extracellulaire, notamment avec la fibronectine (Singer et coll., 1984). Les composants de la matrice extracellulaire jouent un rôle prépondérant dans la transformation phénotypique des fibroblastes. Ainsi, il a récemment été montré dans un modčle expérimental de fibrose hépatique que les modifications de la matrice extracellulaire précédaient la différenciation
myofibroblastique (Desmouliere et coll., 1997). De męme, la fibronectine comportant le domaine EIIIA, exprimée par les cellules endothéliales au cours de la cicatrisation, est capable d’induire la transformation en myofibroblastes de cellules périsinusoďdales (Jarnagin et coll., 1994).
Parmi les cytokines étudiées, certaines, comme le TGFb-1 ou -2, l’endotheline-1, le PDGF, induisent cette différenciation tandis que d’autres, telles le TGF-b3 et l’interféron-g, l’inhibent (Monte Alto Costa, 1995).
En plus des fonctions déjŕ attribuées aux fibroblastes, les myofibroblastes seraient également responsables des forces provoquant la contraction des plaies (cf II C.2.).
Récemment, un nouveau type cellulaire d'origine sanguine et nommé fibrocyte a été décrit. Son phénotype particulier (collagčne+/vimentine+/CD34+) et sa présence dans les cicatrices suggčrent un rôle actif dans la cicatrisation (Bucala et coll., 1994). Depuis sa découverte, son implication dans un rôle de présentation d'antigčne a pu ętre démontré (Chesney et coll., 1997).
Ce processus correspondant ŕ la formation de nouveaux vaisseaux sanguins permet d’apporter au tissu de granulation en formation, et ultérieurement en remodelage, les apports nécessaires en oxygčne et en nutriments. Concommitante ŕ la formation du tissu cicatriciel, l’angiogénčse est sous la dépendance étroite des produits de l’inflammation, de la fibroplasie et du remodelage tissulaire. Les différents mécanismes conduisant ŕ la formation des nouveaux vaisseaux commencent ŕ ętre établis.
Les monocytes/macrophages recrutés sur le foyer de blessure par les fragments de fibronectine et d’autres produits de dégradation de la matrice extracellulaire libčrent différents facteurs dont le b-FGF et le VEGF, capables de stimuler la libération par les cellules endothéliales d’activateur du plasminogčne et de protéinases, qui peuvent ensemble, dégrader les différents constituants de la membrane basale. Cette fragmentation permet la transmigration des cellules endothéliales et leur migration vers le site de la blessure en réponse aux facteurs angiogéniques.
Les cellules endothéliales migrent les unes derričre les autres et forment un bourgeon solide. Cette migration est favorisée par de nombreuses substances chimioattractantes comme le TNF-a, la fibronectine, le b-FGF, le VEGF et l’héparine libérée par les mastocytes. Les cellules situées derričre ce bourgeon constituent la population proliférante. Cette prolifération semble médiée par divers stimulants interagissant entre eux, dont des facteurs solubles ŕ action autocrine ou paracrine, la composition et l’organisation de la matrice extracellulaire environnante et les interactions cellule-cellule (Madri, 1996).
Au fur et mesure de leur élongation et de leur migration ŕ travers la blessure, les bourgeons endothéliaux forment des tubes creux permettant le rétablissement de la circulation sanguine. Les mécanismes qui président ŕ la formation de ces tubes restent encore incertains mais semblent impliquer la création d’espace entre les membranes plasmiques de cellules adjacentes qui seraient jointes par des molécules d’interactions homotypiques, telle PECAM-1 (Albelda et Buck, 1990). Cette structure serait alors stabilisée par des interactions avec la matrice extracellulaire via des intégrines (sous-unité b1) et la formation de jonctions serrées (Merwin et coll., 1990, Merwin et coll., 1991).
Aprčs apposition et inclusion des péricytes dans la membrane basale des capillaires, des fibroblastes viennent s'accoler et se différencient en cellules musculaires lisses. Celles-ci sécrčtent des enzymes induisant l'activation du TGF-b qui a un effet inhibiteur sur la migration et la prolifération des cellules endothéliales. Les cellules endothéliales, fibres musculaires lisses et péricytes sécrčtent et forment alors une membrane basale (Sephel et coll., 1996). Aprčs l’arręt de la stimulation angiogénique, la vascularisation subit un profond remodelage et une involution caractérisée par une adhésion plaquettaire aux cellules endothéliales, une stase sanguine, une apoptose des cellules endothéliales et la digestion des débris cellulaires par les macrophages locaux (Clark, 1995, Pollman et coll., 1999). Récemment, une implication de la métalloélastase a également été suggérée dans ce phénomčne (Madlener et coll., 1998).
La derničre phase de la cicatrisation est constituée par le remodelage de la matrice extracellulaire, la maturation des fibres et l’apoptose cellulaire qui aboutit ŕ la formation d’un tissu paucicellulaire et résistant proche du tissu conjonctif initial. La phase de remodelage tissulaire recouvre celle de la formation du tissu de granulation. Elle permet le passage du caillot de fibrine ŕ une matrice extracellulaire immature principalement constituée de fibronectine et d’acide hyaluronique puis ŕ un tissu riche en collagčne, de type III dans un premier temps puis de type I, et en protéoglycannes. Au niveau spatial, les différentes matrices commencent par ętre déposées prčs des berges de la plaie puis se développent vers le centre de la blessure.
L’évolution du tissu cicatriciel dans le temps et l’espace dépend des cellules qui produisent les macromolécules qui le constituent, des cytokines qui modulent les comportements cellulaires, des protéinases qui permettent de passer d’une matrice ŕ l’autre et des matrices extracellulaires elles-męmes qui participent ŕ ce systčme d’interactions et de rétrocontrôle cellule-cytokines-matrice déjŕ évoqué sous la notion de réciprocité dynamique.
Seront évoqués ici la nature et les rôles des différentes matrices extracellulaires qui se succčdent pour aboutir au tissu cicatriciel mature ainsi que les différentes cytokines qui peuvent intervenir dans leur formation. Le mécanisme permettant le passage d’une matrice ŕ l’autre implique principalement les protéinases et sera détaillé dans la partie suivante.
Le caillot de fibrine constitue la premičre matrice extracellulaire déposée dans l’espace de la blessure. Cette fonction pourrait également ętre exercée par les fragments dénaturés de collagčnes. Le caillot de fibrine contient en fait de nombreuses protéines, parmi lesquelles la fibronectine, la vitronectine, le facteur de von Willebrand, la thrombospondine, le plasminogčne, les activateurs du plasminogčne et leurs inhibiteurs, la thrombine, l’a2-macroglobuline ainsi que différentes cytokines.
Le rôle de ce caillot de fibrine, outre celui lié ŕ la coagulation, est de fournir une matrice provisoire sur laquelle les cellules peuvent adhérer et migrer (Brown et coll., 1993b). Nous avons déjŕ évoqué plus avant l’implication de la fibronectine, de la vitronectine et des récepteurs de type intégrine dans ces mécanismes. (cf II.1.1.)
Un autre rôle de cette matrice est celui de réservoir de facteurs de croissance. Ainsi, il a été démontré que le PDGF et le TGF-b peuvent se lier ŕ la fibrine (Yamada et Clark, 1996). Il est d’ailleurs intéressant de noter que la réponse des fibroblastes ŕ ces facteurs semble influencée par la nature de la matrice extracellulaire dans laquelle ils sont localisés. Ainsi, des fibroblastes répondent davantage au signal de synthčse de collagčne envoyé par le TGF-b lorsqu’ils sont situés dans une matrice de fibrine que dans ŕ une matrice de collagčne (Clark et coll., 1995). Ceci suggčre que la matrice de fibrine permet davantage l’expression d’un phénotype fibroblastique de tissu de granulation tandis qu’un environnement collagénique promeut celui de fibroblastes résidents dermiques.
L’invasion du caillot de fibrine par le tissu de granulation implique la lyse du caillot. Cette dégradation passe par l’expression de protéinases par les fibroblastes localisés sur les berges de la plaie.
Dans le męme temps oů ils dégradent le caillot de fibrine, les fibroblastes forment une nouvelle matrice extracellulaire constituée de fibronectine et d’acide hyaluronique. Il y a donc passage d’une matrice provisoire d’origine plasmatique ŕ une matrice provisoire produite in situ par les cellules, la rendant ainsi plus modelable.
La fibronectine est une protéine d’adhésion trouvée ŕ la fois dans le plasma (ce qui explique sa présence dans le caillot de fibrine) et dans les tissus. Bien qu’elle soit codée par un seul gčne, la fibronectine possčde de nombreuses isoformes provenant d’un épissage alternatif. Ainsi, sa masse moléculaire varie entre 250 et 350 kDa ; toutefois, elle est toujours composée de deux chaînes identiques liées par un pont disulfure (Johansson, 1996). Différentes formes de fibronectine sont retrouvées dans les blessures, mais leur rôle respectif n’est pas encore clairement établi.
La structure de la fibronectine permet l’adhésion et la migration de différents types cellulaires, en particulier les fibroblastes, les kératinocytes et les cellules endothéliales (Brown et coll., 1993a). A travers un fragment de 120 kDa contenant la séquence tripeptidique RGD, elle possčde également une activité chimiotactique pour les fibroblastes, les kératinocytes et les monocytes (Postlethwaite et coll., 1981, Bowersox et Sorgente, 1982). La fibronectine intervient aussi dans la détersion de la blessure en permettant l’opsonisation de fragments de la matrice extracellulaire, stimulant ainsi leur phagocytose par les macrophages (Brown et Goodwin, 1988).
L’acide hyaluronique est un polymčre linéaire de séquences dissaccharidiques [N-acétylglucosamine-acide glucuronique]n. C’est un glycosaminoglycanne produit par la plupart des cellules et particuličrement abondant autour des cellules mésenchymateuses. C’est le composé majeur du tissu de granulation précoce (Clark 1995).
Comme la fibronectine, l’acide hyaluronique permet l’adhésion et la migration de nombreux types cellulaires. Différents mécanismes, non mutuellement exclusifs, sont avancés. Ainsi, l’acide hyaluronique pourrait faciliter l’adhésion-désadhésion entre les héparannes sulfates de la surface cellulaire et la fibronectine de la matrice extracellulaire (Clark, 1995). D’autres hypothčses impliquent des récepteurs protéiques de l’acide hyaluronique situés ŕ la surface de cellules comme les fibroblastes ou les macrophages. Deux de ces récepteurs sont déjŕ caractérisés : le CD44 et RHAMM (Receptor for Hyaluronic Acid Mediated Motility) (Hardwick et coll., 1992, Turley, 1992).
D’autres études suggčrent la capacité de l’acide hyaluronique ŕ stimuler la prolifération cellulaire des fibroblastes ou des cellules épidermiques (Greco et coll., 1998).
La fibronectine et l’acide hyaluronique présents précocement dans le tissu de granulation sont progressivement remplacés par les collagčnes et les protéoglycannes qui sont des substances mécaniquement plus résistantes. La fibronectine est dégradée par les protéinases cellulaires ou plasmatiques ; l’acide hyaluronique l’est par une hyaluronidase tissulaire, enzyme possédant une activité endoglycosidasique spécifique (Bertolami et Donoff, 1982).
Deux types de collagčnes fibrillaires sont majoritairement détectés dans le tissu cicatriciel : le collagčne de type III dans les étapes précoces puis, le collagčne de type I qui le remplace progressivement. Le dépôt de collagčne débute dčs les premiers jours puis atteint un maximum ŕ trois semaines. La synthčse se poursuit aprčs cette date, mais le dépôt n’augmente plus car il s’opčre un équilibre entre synthčse et dégradation. Différentes expériences d’hybridation in situ ont permis de montrer que l’expression d’ARNm des chaînes a1(I) est d’abord détectée dans les couches profondes du tissu de granulation, puis du 6čme au 13čme jour, un taux croissant de transcrit est localisé au niveau des fibroblastes des couches supérieures. Du 13čme au 23čme jour, la synthčse diminue dans les couches profondes tandis qu’une forte expression est visible dans les couches subépidermiques. Aprčs 26 jours, le marquage devient trčs faible et est restreint aux couches supérieures (Oono et coll., 1993).
L’expression des autres types de collagčne est moins connue, toutefois on sait que le collagčne de type V augmente dans le tissu de granulation parallčlement ŕ la néovascularisation et que le collagčne de type VI est exprimé pendant la cicatrisation par les fibroblastes et les cellules endothéliales des néovaisseaux. Son expression permettrait de fournir un ancrage aux néovaisseaux (Oono et coll., 1993).
En plus de leur rôle de support et de résistance, les collagčnes peuvent avoir des effets sur les cellules et sur la matrice extracellulaire. Ainsi, certains peptides provenant des collagčnes sont chimioattractants pour les fibroblastes in vitro et in vivo. Les collagčnes intacts peuvent également moduler le phénotype et la fonction de différents types cellulaires via des récepteurs de type intégrines a1b1 et a2b1 (Clark, 1985, Borel, 1998). La prolifération et la synthčse de collagčne par des fibroblastes cultivés dans une matrice de collagčne est, par exemple, diminuée (Gillery et coll., 1992b) alors que l’expression d’intégrine a2b1 et de collagénase est augmentée (Klein et coll., 1991). La matrice extracellulaire riche en collagčnes qui s’accumule dans le tissu de granulation mature diminue la capacité des fibroblastes ŕ produire davantage de matrice collagénique mais augmente leur capacité ŕ remodeler cette matrice.
Les protéoglycannes agissent, eux aussi, dčs les premiers temps de la cicatrisation, en particulier dans la régulation de la coagulation. Ils interviennent aussi directement dans les phases plus tardives de la formation du tissu de granulation. Les protéoglycannes ŕ chondroďtines et ŕ dermatanne sulfates sont en effet impliqués dans la fibrillogénčse du collagčne : la décorine en se fixant au collagčne de type I limite la taille des fibres (Vogel et coll., 1984, Danielson et coll., 1997).
Les protéoglycannes peuvent également agir sur les fonctions cellulaires : le versicanne est capable de stimuler la migration cellulaire en diminuant l’adhésion des cellules ŕ leur substrat (Yamagata et coll., 1993). L’apparition des protéoglycannes ŕ chondroďtines sulfates étant concomitante avec une diminution de la prolifération cellulaire, un rôle modulateur de la prolifération est aussi évoqué (Clark, 1995).
L’interaction de ces molécules avec les facteurs de croissance constitue une autre voie d’intervention des protéoglycannes dans la formation du tissu cicatriciel. Différentes études ont déjŕ démontré la capacité des chaînes de GAGs ou des protéines cśur ŕ lier des facteurs de croissance. La protéine cśur de la décorine fixe le TGF-b (Hildebrand et coll., 1994), les chaînes d'héparanne-sulfate fixent les FGF-1, -2, -4, -7, le PDGF, le GM-CSF, l’IL-3, le TGF-b, le PF-4, le VEGF (Clark, 1995). La liaison de ces médiateurs aux protéoglycannes est parfois impliquée dans des phénomčnes de reconnaissance/présentation au récepteur permettant ainsi de moduler le comportement cellulaire. La famille des syndécannes, présents ŕ la surface cellulaire, semble avoir ces fonctions. En outre, la liaison des chaînes de GAGs aux différentes macromolécules de la matrice extracellulaire peut aussi médier les mouvements cellulaires ou permettre la compartimentation des cellules dans un tissu. Ainsi, les syndécannes-1 et -4 facilitent la formation de contacts focaux dans les fibroblastes (Gallo et coll., 1996).
Il est ŕ noter que l’élastine, dont le rôle est essentiel pour conférer son élasticité ŕ la peau, n’est réexprimée que faiblement et trčs tardivement dans la blessure, ce qui explique la rigidité du tissu cicatriciel.
La contraction du tissu cicatriciel qui survient ŕ partir du 6čme jour est essentiellement dűe aux myofibroblastes (Monte Alto Costa, 1995). Déjŕ décrits au paragraphe II.B.3.1., les myofibroblastes exercent des forces de traction sur la matrice aboutissant ŕ la compaction du tissu et la contraction de la blessure. Il est ŕ noter qu’ŕ l’inverse des capillaires ou des macrophages, les myofibroblastes s’alignent dans le tissu de granulation mature le long des lignes de contraction.
Outre ses aspects biomécaniques, il semble que ce processus de rétraction nécessite l’intervention d’un signal. L’expression de PDGF BB ou AB par les macrophages pourrait fournir ce signal ; l’observation d’un second pic d’expression d’ARNm de la chaîne B du PDGF aprčs 6 jours de culture in vitro de monocytes sur une matrice de fibronectine ou de collagčne va dans ce sens (Shaw et coll., 1990).
Pendant les trois premičres semaines, une cicatrice possčde environ 20% seulement de sa résistance finale ŕ la déchirure. Si ce taux est directement lié ŕ l’accumulation de collagčne et ŕ la contraction de la blessure, l’augmentation progressive de la résistance qui se produit ensuite est davantage due ŕ un remodelage du tissu permettant la formation de plus gros amas de collagčne et ŕ une maturation des liaisons croisées intermoléculaires.
La notion de remodelage tissulaire renvoie aux modifications transitoires ou permanentes de l’architecture du tissu cicatriciel. Ce remodelage se produit lors des différentes phases de la réparation tissulaire (inflammation, formation du tissu de granulation et du tissu mature) et implique la production et l'activation de protéinases par les cellules du tissu conjonctif. La matrice extracellulaire étant constituée d’une grande variété de macromolécules, les protéinases impliquées dans leur dégradation sont, elles aussi, trčs nombreuses. On distingue trois grandes classes de protéinases : les sérine protéinases, les métalloprotéinases matricielles (MMPs) et les cystéines protéinases.
Parmi ces protéinases, les sérines protéinases et les MMPs semblent les plus impliquées dans le remodelage tissulaire. Si les MMPs, plus spécifiquement étudiées dans ce travail seront détaillées, il convient de noter la place importante prise par les activateurs du plasminogčne dans le remodelage cicatriciel.
Les activateurs du plasminogčne de type urokinase (uPA) ou tissulaire (tPA), forment avec l’élastase leucocytaire et la cathepsine G, l’essentiel de la famille des sérines protéinases. Catalyseurs de la formation de plasmine ŕ partir du plasminogčne, ils ont d’abord été décrits comme responsable de la dégradation du caillot de fibrine. La faible spécificité de la plasmine, capable de cliver de nombreuses protéines, permet en fait d’étendre le rôle des activateurs du plasminogčne dans le remodelage tissulaire ŕ la dégradation de la matrice extracellulaire, ŕ la libération et ŕ l’activation de facteurs de croissance, ŕ la migration des cellules, ŕ l’angiogénčse et ŕ la réépithélialisation (Mignatti, 1996).
Les MMPs représentent une famille d’enzymes possédant les caractéristiques suivantes :
Selon leur spécificité et leur substrat préférentiel, les MMPs peuvent ętre classées en au moins 5 groupes distincts : collagénases, gélatinases, stromélysines, métallo-élastases, MMPs de type membranaire (MT-MMPs : Membrane type Matrix metalloproteinases) (Tableau VI).
Les collagénases (MMP-1, MMP-8, MMP-13, MMP-20) initient spécifiquement la dégradation des collagčnes fibrillaires (de type I, II et III). Elles clivent la triple hélice pour donner deux fragments correspondant aux 3/4 N-terminaux et 1/4 C-terminaux. Cette coupure provoque le déroulement de la triple hélice et libčre un collagčne dénaturé, alors accessible aux protéinases non spécifiques.
La spécificité de ces collagénases est variable : la collagénase-1 (MMP-1 ou collagénase interstitielle) dégrade le collagčne de type III plus efficacement que les types I et II tandis que la MMP-8 (ou collagénase-4) est plus active sur le type I. La collagénase-3 (MMP-13) possčde une spécificité de substrat plus large : outre les collagčnes fibrillaires et la gélatine, elle dégrade aussi les collagčne de type IV, IX, X et XIV. Cette collagénase possčde également un site de clivage supplémentaire dans le collagčne de type I au niveau du domaine non collagénique N-terminal. Il est ici important de noter que la MMP-13 est la collagénase prédominante chez les rongeurs (Quinn et coll., 1990), qui ne possčdent pas d’homologues de la MMP-1 humaine.
Les gélatinases ou collagénases de type IV sont classées en deux types : la gélatinase A ou gélatinase de 72 kDa (MMP-2) et la gélatinase B ou gélatinase de 92 kDa (MMP-9). Ces deux enzymes dégradent principalement les collagčnes dénaturés et le collagčne de type IV. Toutefois, des études récentes leur attribuent également des activités collagénolytiques. Ainsi, la MMP-2 est capable de cliver le collagčne de type I en fragments 3/4, 1/4 identiques ŕ ceux générés par les collagénases (Aimes et Quigley, 1995). La MMP-9 exprimée par les ostéoclastes durant le développement foetal de la souris est, elle, capable de cliver les collagčnes de type I, II et V dans le télopeptide N-terminal (Kahari et Saarialho-Kere, 1997a).
Les Stromélysines possčdent une spécificité de substrat trčs large. Les stromélysines-1 (MMP-3) et -2 (MMP-10) sont capables de dégrader les protéines coeur des protéoglycannes, les laminines, la fibronectine, l’élastine, le nidogčne, la gélatine, les collagčnes de type IV, V, IX et X. La stromélysine-3 (MMP-11), et c’est une exception, ne possčde pas de substrat connu au sein de la matrice extracellulaire mais dégrade un inhibiteur de sérine-protéinase.
Les MMPs de type membranaire (MT-MMPs) forment un sous-groupe d’au moins 4 isoformes. La spécificité de substrat de la MT1-MMP (MMP-14) est la mieux connue. Si des études récentes ont montré qu’elle pouvait dégrader certains composés de la matrice extracellulaire (Kahari et Saarialho-Kere, 1997a, Ohuchi et coll., 1997), cette activité semble faible (D'Ortho et coll., 1998). Les MT1-MMP, MT2-MMP et MT3-MMP sont également décrits comme activateurs physiologiques de la MMP-2. Il est également ŕ noter qu'une forme soluble de la MT3-MMP a été décrite chez l'homme (Matsumoto et coll., 1997). Provenant d'un épissage différentiel du gčne codant la MT3-MMP, cette forme est capable d'activer la proMMP-2 mais aussi de dégrader différents substrats matriciels (fibronectine, collagčne de type III).
La métallo-élastase (MMP-12) dégrade préférentiellement l’élastine mais également la fibronectine.
La matrilysine (MMP-7) possčde un spectre de dégradation trčs large : fibronectine, laminine, entactine, collagčne de type IV et protéine-coeur des protéoglycannes.
Récemment, de nouveaux membres de la famille des gčnes des MMPs ont été clonés et désignés comme MMP-18 (Cossins et coll., 1996) et MMP-19 (Pendas et coll., 1997) mais leur spécificité de substrat n’est pas encore connue. La MMP-20 est une nouvelle métalloprotéinase ŕ activité collagénolytique isolée ŕ partir d'une banque d'ADNc d'odontoblastes (Llano et coll., 1997).
La structure primaire des MMPs contient cinq domaines fonctionnels :
Il est ŕ noter que la MMP-7 fait exception car elle ne possčde dans sa structure que les trois premiers domaines parmi les cinq décrits ci-dessus (Figure 11) (Ray et Stetler-Stevenson, 1994).Les gélatinases contiennent un domaine supplémentaire en aval du domaine de liaison du Zinc. Ce domaine, dit fibronectin-like en raison de son homologie avec la protéine du męme nom, est responsable de la forte affinité de ces enzymes pour le collagčne et la gélatine (Murphy et coll., 1994, Collier et coll., 1992). La MMP-9 et les MT-MMPs contiennent également un domaine homologue ŕ la chaîne a2 du collagčne de type V (Wilhelm et coll., 1989). Les MT-MMPs possčdent un domaine additionnel ŕ l’intérieur du domaine catalytique qui permet leur ancrage membranaire (Sato et coll., 1994). En outre, elles partagent avec la stromélysine-3 une séquence de 10 résidus située entre le prodomaine et le domaine catalytique qui est homologue aux séquences de reconnaissance des enzymes furin-like (Sato et coll., 1996).
L’absence d’activité enzymatique des formes zymogčnes résulte de l’interaction entre le Zinc du site actif et un résidu Cystéine présent dans le prodomaine de la molécule. L’activation enzymatique correspond ŕ la dissociation Cys-Zn2+ et ŕ la fixation d’une molécule d’eau sur l’atome de Zinc, suivie d’une auto-protéolyse qui libčre le pro-fragment N terminal, rendant ainsi le site actif accessible. La rupture de cette interaction Cys-Zn2+ ("Cystein switch" dans la terminologie anglo-saxonne) peut se produire sous l’influence d’agents chaotropiques tels que les organomercuriels, par des agents physiques comme la chaleur (Koklitis et coll., 1991) ou par clivage protéolytique du propeptide (Figure 12). Ainsi, plusieurs sérines protéinases, dont la plasmine, la trypsine, la chymase des mastocytes peuvent activer certaines MMPs (Okada et Nakanishi 1989, Rice et Banda, 1995).
Bien que les mécanismes d’activation des MMPs in vivo ne soient pas encore totalement élucidés, quatre voies d’activation sont possibles:
(1) L’activation extracellulaire des MMPs par des protéinases “non-MMPs”. Elle fait intervenir la cascade de la plasmine et peut se produire ŕ la surface cellulaire via le récepteur de l’uPA ou ętre distant du site de sécrétion de l’enzyme (He et coll., 1989). Dans ce systčme, le plasminogčne est clivé en plasmine par l’uPA ou le tPA. La plasmine ainsi formée peut alors activer totalement la stromélysine et partiellement la collagénase interstitielle. La stromélysine active peut augmenter l’activité de la collagénase interstitielle par un clivage d’un peptide de l’extrémité C terminale de la molécule. Un autre exemple de ce type d’activation est fourni par la thrombine capable d’activer la MMP-2 (Galis et coll., 1997, Zucker et coll., 1995).
(2) L’activation extracellulaire des MMPs par d’autres MMPs. La matrilysine a ainsi été décrite comme étant capable d’activer les proMMP-2 , -3 et -9. La proMMP-9 peut également ętre activée par la MMP-2 (Fridman et coll., 1995).
(3) L’activation intracellulaire par la furine. La furine est une sérine protéinase du réseau trans de l’appareil de Golgi qui reconnaît la séquence RXKR portée par les stromélysine -1, -2, -3 et les MT-MMPs. Cependant, ce type d’activation n’a été montré que pour la prostromélysine-3 et la MT1-MMP (Sato et coll., 1996).
(4) L’activation par les MT-MMPs. Elle semble ne concerner que la proMMP-2. A l’inverse des autres MMPs, l’activation de la proMMP-2 se produit ŕ la surface cellulaire et fait intervenir les MT-MMPs. Le rôle des MT-MMPs n’est pas totalement clair et fait encore l’objet de nombreux travaux męme si un rôle de récepteur membranaire de la pro-enzyme est couramment admis (Schwartz et coll., 1998). Actuellement, il semble que le systčme d’activation de la proMMP-2 ferait intervenir un complexe tertiaire entre la MT-MMP, le TIMP-2 et la proMMP-2 (Emonard et coll., 1992, Strongin et coll., 1995, Kinoshita et coll., 1998).
La dégradation de la matrice extracellulaire par les MMPs peut ętre régulée ŕ différents niveaux : transcription des gčnes codant les MMPs, stabilisation des ARNm, traduction, sécrétion sous forme zymogčne, liaison des proenzymes ŕ la membrane et/ou aux molécules de la matrice extracellulaire, activation, inhibition par les TIMPs, dégradation.
En rčgle générale, les MMPs ne sont pas exprimées constitutivement in vivo mais sont induites en réponse ŕ des cytokines, des facteurs de croissance, des hormones. Ainsi, la plupart des MMPs sont induites par l’IL-1 b, le TNF-a, le PDGF, le TGF-a, l’EGF, le bFGF et le NGF (Ray et Stetler-Stevenson, 1994). Ces effets s’expliquent en partie par la présence dans la région 5’-flanquante de la partie codante du gčne de séquences cis-régulatrices de type AP-1 et/ou PEA3. Ces séquences stimulent la transcription du gčne situé en aval lorsqu’elles sont liées au facteur trans-régulateur correspondant (complexe jun/fos pour le site AP-1 et c-ets pour le site PEA3) (Mauviel, 1993). L’activation AP-1-dépendante des MMPs peut ętre inhibée par les glucocorticoďdes ou l’acide rétinoďque (Kahari et Saarialho-Kere, 1997a).
A l’inverse des autres MMPs, la MMP-2 est exprimée constitutivement par de nombreux types cellulaires. L’organisation de la séquence promotrice du gčne montre des différences notables avec celle des autres MMPs. Ce promoteur contient une boite TATA non canonique et ne possčde ni site AP-1 ni site PEA3 mais deux sites SP1 et un site de liaison ŕ la p53 (Corcoran et coll., 1996, Bian et Sun, 1997).
La régulation de l’expression des MMPs est spécifique du type cellulaire et certains facteurs peuvent avoir des effets opposés sur des types cellulaires différents. C’est par exemple le cas du TGF b qui stimule la production de MMPs par les kératinocytes mais inhibe leur expression dans les fibroblastes (Salo et coll., 1991, Edwards et coll., 1987).
D’autre part, l’expression des MMPs est dépendante de l'environnement péricellulaire, des interactions cellule-cellule et cellules-matrice extracellulaire ainsi que de la forme de la cellule (Werb et coll., 1986, Seftor et coll., 1993).
La famille des TIMPs comprend au moins 4 isoformes chez le rat désignées sous les noms de TIMP-1, TIMP-2, TIMP-3, TIMP-4 , de masses moléculaires respectives 30, 20, 22 et 22 kDa. Exprimés par de trčs nombreux types cellulaires, ils sont présents dans la majorité des tissus et des fluides de l’organisme. Si les TIMPs-1 et -2 sont présents sous forme soluble, le TIMP-3 est insoluble et lié ŕ la matrice extracellulaire (Leco et coll., 1994). Le TIMP-1 se distingue également par la forte glycosylation de sa chaine polypeptidique constitutive, faisant passer la masse moléculaire de 20 kDa ŕ 30 kDa (Charvat, 1995). Une quatričme isoforme (TIMP-4) récemment découverte par clonage chez l’homme et la souris semble n’ętre significativement exprimée que dans le coeur (Leco et coll., 1997).
Ces inhibiteurs partagent une forte homologie de structure (entre 37 et 51%). Ainsi, ils comportent 12 résidus de cystéine permettant la formation de 6 ponts disulfure qui organisent la molécule en deux domaines et sont responsables de la formation des boucles impliquées dans la liaison avec l’enzyme. Ils forment avec les MMPs des complexes équimolaires non covalents de haute affinité (Gomez et coll., 1997). Plusieurs modes d’interactions entre TIMPs et MMPs semblent exister :
Le rôle anti-protéinasique des TIMPs peut s’exercer ŕ plusieurs niveaux :
Il convient ici de noter que si les TIMPs inhibent spécifiquement les MMPs, il existe également des inhibiteurs non spécifiques tels que l’a2- macroglobuline et d’a1-protéinase, capables de diminuer l'activité de ces protéases (Grinnell et coll., 1998).
Dans la cicatrisation, les collagénases sont principalement produites par les kératinocytes basaux de l’épiderme. Différentes études immunohistologiques ont en effet montré une expression de la collagénase interstitielle au niveau des kératinocytes migrants qui sont au contact des composants de la matrice extracellulaire (Inoue et coll., 1995, Saarialho-Kere et coll., 1993). Cette interaction entre les kératinocytes et la matrice dermique, en particulier avec le collagčne de type I, pourrait fournir un signal précoce d’initiation de la réponse épithéliale ŕ la blessure. D’autres études réalisées in vivo ou in vitro ont permis de montrer que cette interaction impliquait des récepteurs de type intégrine exprimés constitutivement (a1b1; a2b1; a3b1; a6b4) ou sélectivement (a5b1; avb3) par les cellules migrantes. La production de collagénase par ces cellules pourrait aussi ętre influencée par des facteurs solubles (IL-1, TGF-a) présents dans l’environnement de la blessure ou produits par l’épiderme (Turksen et coll., 1991,Mauviel, 1993)
Certains travaux ont également détecté une expression de collagénase dans le derme superficiel au niveau de cellules mésenchymateuses et inflammatoires (Madlener et coll., 1998, Saarialho-Kere et coll., 1992). Toutefois, ces signaux sont plus épars et moins intenses que leur équivalents épidermiques. In vitro, les fibroblastes sont capables de synthétiser de la collagénase interstitielle; cette synthčse peut ętre stimulée par des cytokines comme IL-1, TNF-a, PDGF, EGF et par une interaction des cellules avec le collagčne de type I, comme cela a pu ętre démontré en les cultivant dans un lattis de collagčne (Mauch et coll., 1989).
L’activité de dégradation des collagénases pourrait ętre impliquée dans différentes étapes de la cicatrisation, en fonction du compartiment dans lequel elle s’exprime et/ou agit. Ainsi, dans l’épiderme, le rôle des collagénases serait essentiellement de promouvoir la migration cellulaire. La dégradation du collagčne de type I permettrait aux kératinocytes de se dissocier localement de leur substratum et de migrer pour reformer un néo-épithélium. En revanche, l’activité collagénolytique du derme permettrait le remodelage matriciel du tissu de granulation.
Les différentes isoformes des stromélysines ont été détectées lors de la cicatrisation de blessures aiguës et chroniques. Bien que ces enzymes possčdent une large spécificité de substrats et une organisation protéique similaire, leur localisation au sein du tissu est trčs différente.
Ainsi, la stromélysine-2 est uniquement produite par les kératinocytes basaux situés au front de migration de l’épithélium. Les kératinocytes en question sont les męmes que ceux exprimant la collagénase interstitielle.
La stromélysine-1 est également produite ŕ ce niveau mais par une population de kératinocytes différents, dans des cellules proliférantes, “derričre” le front de migration. Ces cellules sont encore au contact des différents composants de la membrane basale. D’autre part, un fort signal pour l’expression de la stromélysine-1 est également détecté au niveau des fibroblastes du tissu de granulation (Saarialho-Kere et coll., 1994, Madlener et coll., 1998).
Le profil d’expression de la stromélysine-3 a été étudié chez le rat dans la cicatrisation dermique de blessures aiguës et montre une localisation des ARNm confinée dans le tissu stromal; on note une co-localisation avec les ARNm de la MMP-2 et de la MT1-MMP (Okada et coll., 1997).
L’expression de ces stromélysines peut ętre modulée par les cytokines présentes dans le tissu. Des études faites in vitro ont ainsi montré que l’EGF, le KGF et le TNF-a stimulent l’expression de stromélysine-2 tandis que l’IL-1, le PDGF, l’EGF, le TNF-a augmentent celle de la stromélysine-1 par les fibroblastes ou les kératinocytes (Madlener et coll., 1996).
A la différence des collagénases qui possčdent un spectre d’action limité aux collagčnes fibrillaires (type I, II, III), les stromélysines sont capables de dégrader de nombreuses protéines de la matrice extracellulaire : fibronectine, laminine, collagčne de type IV, GAGs. Cette grande diversité de substrat et la localisation tissulaire de ces enzymes laissent penser ŕ un rôle de la stromélysine-2 dans la migration des kératinocytes via la dégradation des molécules de la membrane basale. Un rôle d’activateur de la MMP-9 est aussi envisagé. Localisée dans les cellules proliférantes, la stromélysine-1 synthétisée par les kératinocytes basaux de l’épiderme semblerait davantage impliquée dans les remodelages tissulaires précédant la formation d’une nouvelle membrane basale. Dans le derme, les stromélysines -1 et -3 peuvent ętre impliquées dans la formation et le remodelage du tissu de granulation et/ou dans des processus qui y sont associés tels l’angiogénčse, l’extravasation et la migration de cellules inflammatoires (Mignatti, 1996).
L’étude de l’expression des gélatinases pendant la cicatrisation cutanée montre de grandes différences spatiales et temporelles en fonction de l’isoforme considérée.
La MMP-9 semble impliquée dans les phénomčnes précoces de la réparation : son expression augmente dčs le premier jour aprčs la blessure et diminue rapidement. Cette expression, qui coďncide avec la période de détersion, est associée ŕ la migration dans le foyer de blessure des cellules inflammatoires qui la sécrčtent : neutrophiles, éosinophiles, macrophages. Des études d’hybridation in situ montrent également une synthčse de MMP-9 au niveau des régions proliférantes de l’épithélium et plus tardivement, au niveau du tissu de granulation. Ces localisations peuvent s’expliquer par l’expression de l’enzyme par les kératinocytes et les cellules endothéliales capillaires (Madlener et coll., 1998).
Si la MMP-2 est exprimée constitutivement, sa production augmente aprčs une blessure. Son profil d'expression diffčre des autres MMPs, avec un pic plus tardif et surtout une expression exclusivement mésenchymateuse, pendant la formation du tissu de granulation.
Récemment, des études ont permis de montrer que l’expression de MMP-2 dans ce tissu était co-localisée avec celle de la MT1-MMP (Okada et coll., 1997, Madlener et coll., 1996).
L’ensemble de ces données permettent d’envisager des rôles distincts pour ces deux enzymes. La MMP-9 serait davantage impliquée dans les étapes plus précoces telles que le détachement des kératinocytes de la membrane basale, la migration de ces cellules et le remodelage des matrices provisoires (fibrine-fibronectine) (Salo et coll., 1994). La MMP-2 parait, pour sa part, agir plus tardivement dans les phases de remodelage de la matrice extracellulaire (Okada et coll., 1997, Kahari, Saarialho-Kere 1997).
Le GHK-Cu utilisé dans cette étude est composé de 2 moles de tripeptide Glycine-L-Histidyl-L-Lysine (GHK) pour une mole de cuivre (Cu2+). Différentes études de résonance paramagnétique électronique, de titration potentiométrique, de spectroscopie d'absorption et de spectroscopie de masse ont permis de montrer, ŕ pH neutre, la formation d'un complexe tertiaire GHK-Cu 2:1. Dans ce complexe, schématisé figure 13, le cuivre est lié aux tripeptides par la fonction NH2 de la glycine, par l'azote impliqué dans la liaison peptidique G-H et par les atomes d'azote des noyaux imidazoles des deux histidines (Lau et Sarkar 1981, Rainer, 1985).
L'affinité du peptide GHK pour le cuivre (Ka=1016.4) est proche de celle de l'albumine (Ka=1016.2). Cette affinité est encore plus élevée (Ka=1022.0) avec un complexe ternaire GHK, Histidine, Cu (Lau et Sarkar, 1971).
Le tripeptide GHK a initialement été isolé dans le plasma lors d'une étude concernant les effets de différentes fractions sériques sur le méFolisme d'hépatocytes de rat en culture. Il est alors apparu que le la forme biologiquement active du tripeptide était celle d'un complexe avec le cuivre (Pickart et Thaler 1973, Pickart et Thaler, 1980, Schlesinger et coll., 1977). L'origine endogčne n'est encore qu'hypothétique; toutefois, des travaux ont montrés que le tripeptide pouvait ętre libéré ŕ partir de protéines de la matrice extracellulaire par un clivage par des protéinases telles la plasmine (Lane et Sage, 1994).
Le devenir du GHK-Cu dans l'organisme n'a pas été étudié mais la distribution des complexes du cuivre est connue. Aprčs leur arrivée dans la circulation générale, les ions cuivriques sont immédiatement complexés par des protéines de transport (albumine, transcupréine). Lorsqu'il arrive au niveau du foie, le cuivre, lié d'une maničre trčs lâche ŕ ces complexes, s'en détache pour se fixer sur les récepteurs des membranes des hépatocytes. Il est alors transféré dans le cytosol oů il se fixe sur les métallothionéines (Houot, 1997). La fonction la plus importante du foie dans le domaine du métabolisme du cuivre est la synthčse de la céruloplasmine. Le cuivre, fortement lié ŕ la céruloplasmine, est mis en circulation dans le sang avant de se répartir ensuite dans les différentes tissus dans lesquels il est utilisé aprčs incorporation dans des apoenzymes ou des métalloprotéines (cytochrome-C oxydase, lysyl-oxydase, superoxyde-dismutase) (Lederer, 1987, Kosonen et coll., 1997).
Des études d'injection intraveineuse du tripeptide GHK marqué au tritium ont été réalisées chez la souris et ont permis de montrer que le composé disparaît du plasma avec une demi-vie d'élimination d'environ 16 minutes pendant les deux premičres heures aprčs lesquelles l'élimination semble plus lente. Les męmes études ont également permis d’observer une accumulation préférentielle de GHK dans le rein et le cerveau (Pickart, 1983).
L’activité chimiotactique du tripeptide GHK a été montré sur différents modčles et sur différents types cellulaires. In vitro, Poole et Zetter ont montré que le peptide ŕ 0,2 mM était capable de stimuler la migration de mastocytes péritonéaux de rats dans un gel d’agarose (Poole et Zetter, 1983). En implantant des polymčres (Dacron) contenant 0,25 µg de GHK sous la peau de souris C57/BL, une activité chimiotactique du peptide a été trouvée pour les mastocytes, les polynucléaires neutrophiles et les monocytes/macrophage. (Zetter et coll., 1985).
Un effet identique sur les polynucléaires a également été rapporté pour le cuivre (CuCl2, de 50 ŕ 500 µM) in vitro dans le modčle des chambres de Boyden. Cet effet est précédé par une modification de la forme de la cellule qui se polarise (Hujanen et coll., 1995).
Sur le męme modčle, en utilisant des extraits de cornée préincubés avec du GHK-Cu , Raju et coll. ont montré que le complexe induisait le chimiotactisme de cellules endothéliales (Raju et coll., 1984).
La capacité d’induire la prolifération de cellules différenciées ou d’augmenter leur viabilité en culture furent les premičres propriétés décrites pour le tripeptide. Peu aprčs son isolement ŕ partir du plasma humain en 1973, il fut décrit comme un facteur capable de stimuler la croissance d’une lignée de cellules hépatiques de rat (HTC4) dans un milieu contenant peu de sérum (Pickart et coll., 1973, Pickart et Thaler, 1980). Depuis, il a été décrit comme permettant la survie et le maintien de neurones en culture, la prolifération de cellules néoplasiques humaines (KB, HeLa) (Williams et Cheng, 1980), de cellules folliculaires thyroďdiennes (Ambesi-Impiombato et coll., 1980) et de chondrocytes embryonnaires de poulet (Pesakova et coll., 1995).
D’autre part, l'addition de GHK dans le milieu de culture augmente la viabilité des macrophages, des éosinophiles, des mastocytes et des fibroblastes (Pickart, 1983, Capron et coll., 1978b, Capron et coll., 1978a, Joseph et coll., 1977).
A des concentrations élevées (1 µg/ml), des effets inhibiteurs de la croissance cellulaire ont été rapportés sur les cellules gliales (Pickart, 1983).
Outre son rôle d’inducteur de la mobilisation de l’endothélium déjŕ cité, le complexe GHK-Cu a également été décrit comme facteur angiogénique. Cette propriété a été démontrée ŕ la fois in vitro dans un modčle de formation de cordons endothéliaux (Lane et coll., 1994) et in vivo dans le modčle de la cornée (Raju et coll., 1984, Raju et coll., 1982) ainsi que dans le modčle des biopsies cutanées (Buffoni et coll., 1995).
L’activité SOD-like du complexe fait encore l’objet de controverses. Selon Pickart, le GHK-Cu possčderait une activité SOD-like significative tandis que Miller n’en a détecté aucune sur des fractions microsomales de rats. En revanche, Miller a montré que le GHK-Cu inhibait la peroxydation lipidique catalysée par le fer. Cet effet existe uniquement lorsque le fer provient de la ferritine et procčde d’une inhibition de la libération du fer par son transporteur (Miller et coll., 1990).
L’effet du GHK-Cu sur la synthčse de différents composants de la matrice extracellulaire a été étudié dans notre laboratoire sur des cultures de fibroblastes dermiques et pulmonaires humains normaux. Il a ainsi été montré que le complexe stimulait la synthčse des collagčnes sécrétés dans le milieu de culture d’une façon dose dépendante. Présente dčs 10-12 M, cette stimulation atteint son maximum ŕ 10-9 M et disparaît pour des concentrations plus élevées (10-6 M). Dans les conditions expérimentales utilisées, cet effet se produit sans modification de la prolifération cellulaire (Maquart et coll., 1988, Maquart, 1990).
Un effet stimulant du complexe sur la synthčse de GAGs a également été démontré par notre équipe dans les męmes types cellulaires. Cette stimulation est également dose-dépendante et biphasique. Les concentrations efficaces et optimales sont les męmes que celles trouvées pour la stimulation de la synthčse de collagčnes. L’analyse des types de GAGs produits par les fibroblastes a en outre montré une action préférentielle du GHK-Cu sur l’accumulation des héparannes sulfate dans la couche cellulaire et des dermatannes sulfates dans le milieu de culture (Wegrowski et coll., 1992).
Les premičres études montrant les effets cicatrisants du GHK-Cu ont été réalisées chez le rat au cours des années 80. Ainsi, l’injection réguličre dans des blessure excisionnelles (2 cm de diamčtre) impliquant la totalité de l’épaisseur de l’épiderme ("full thickness") d’une solution contenant 50µg de GHK-Cu accélčre le taux de cicatrisation de façon significative. Les effets les plus prononcés sont observés entre 20 et 25 jours aprčs la blessure (Downey, 1985). Des résultats similaires ont ensuite été trouvés chez la souris, le porc et dans un modčle de brűlures cutanées chez le rat (Pickart, 1987). Ces męmes études ont également montré que la stimulation de la cicatrisation n’est pas retrouvée avec des analogues tels que GHG-Cu, GH-Cu ou K-Cu ni avec le cuivre seul.
Il a également été montré dans un modčle de blessure par biopsie cutanée et par implantation sous-cutanée d’éponges de vinyl que le complexe était capable d’antagoniser les effets inhibiteurs de la cicatrisation induits par la cortisone, cela aussi bien sur la contraction de la plaie que sur le dépôt de collagčne.
Dans notre laboratoire, une étude précédente faite in vivo chez le rat en utilisant le modčle des chambres de blessure (cf. Matériel et méthodes II.1.) a déjŕ été réalisée et a permis de démontrer que le GHK-Cu stimule la production de tissu cicatriciel et plus spécifiquement celle de différents composants de la matrice extracellulaire : collagčne de type I et III, glycosaminoglycannes, élastine, fibronectine. Cette étude a également permis de fixer la dose optimale de GHK-Cu ŕ injecter dans le cylindre ŕ 2 mg/injection. Un effet stimulant du complexe a également pu ętre observé sur l’expression des gčnes codant les collagčnes de type I et III. La spécificité d’action du complexe a aussi été prouvée. En effet, l’injection du tripeptide GHK non complexé au cuivre, du cuivre seul sous forme CuCl2, ou encore d’un tripeptide témoin EHP n’a provoqué aucune stimulation de la synthčse de collagčne dans ce modčle (Maquart et coll., 1993).
Différentes études réalisées chez le rat, le cobaye ou le lapin ont aussi permis d’observer en histologie un effet angiogénique du GHK-Cu sur le tissu cicatriciel et ce dčs les premiers jours (entre 3 et 8 jours) (Buffoni et coll., 1995).
Différentes études ont été réalisées chez l’homme pour tester l’efficacité du GHK-Cu sur la cicatrisation de différents types de blessures cutanées. La premičre d’entre elle, a été réalisée sur un nombre réduit de cas de cicatrisation aiguë de plaies chirurgicales. Les résultats suggéraient un effet positif de la molécule sur la vitesse de fermeture de la plaie (Fish, 1991).
D’autres études faites sur un nombre plus élevé de patients atteints d’ulcčres chroniques du membre inférieur ont montré des résultats positifs en terme de vitesse de fermeture de la plaie, trois fois plus rapide dans le groupe traité par rapport au groupe placebo, et en terme de réduction des surinfections bactériennes de l’ulcčre (7% contre 34%). Ces études ont également précisé que l’obtention des effets positifs du GHK-Cu nécessitait une application immédiate aprčs la détersion de la plaie (Massey, 1992, Mulder, 1994).
Une autre étude réalisée chez le rat a également démontré dans le modčle de blessure excisionnelle linéaire (3,5 cm x 3 cm) que le GHK-Cu augmentait significativement la résistance et la néovascularisation de la plaie (Schmidt, 1994).
Le GHK-Cu est fabriqué commercialisé par une entreprise américaine : Procyte Corporation. Dans le domaine de la cicatrisation, le GHK-Cu est vendu aux Etats-Unis sous différentes formes : une pommade ŕ base de glycérine (Iamin Gel®), un désinfectant (Iamin wound Cleanser®) et des compresses (Osmocyte PCA®) Tous ces produits ont été mis sur le marché pour le traitement des blessures aiguës et chroniques (ulcčre diabétique, ulcčre de jambe, brűlures du premier et deuxičme degré, ulcčre artériel, incision post-chirurgicale, irritation cutanée). Le produit original, Iamin Gel®, commence ŕ ętre commercialisé en Chine, ŕ Taiwan et le sera bientôt dans le reste de l'Asie, en Amérique du Sud et en Afrique du Sud.
Le GHK-Cu commence également ŕ ętre utilisé pour accélérer la cicatrisation aprčs transplantation capillaire ŕ travers une gamme de produits nommé Graftcyte®.
Enfin, depuis juin 1999, Procyte Corporation développe des partenariats avec différentes entreprises et distributeurs afin de commercialiser différents gammes cosmétiques contenant du GHK-Cu (Complex Cu3®, Neova®, Blue Copper®).
On notera qu'en Europe, le GHK est commercialisé par les laboratoires SEDERMA sous forme d'un complexe avec l'acide palmitique sous le nom de "Biopeptide-CL®", pour utilisation dans des produits cosmétiques.
La phase inflammatoire
La phase vasculaire
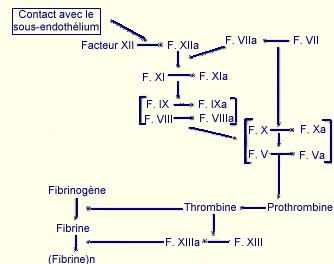
Figure 9La phase cellulaire
Les plaquettes
Les mastocytes
Les polynucléaires neutrophiles
Les macrophages
Les lymphocytes T
La phase proliférative
La réparation épidermique
La migration cellulaire
La prolifération
La maturation
La réparation de la jonction dermo-épidermique
La réparation dermique : formation du tissu de granulation
Fibroplasie
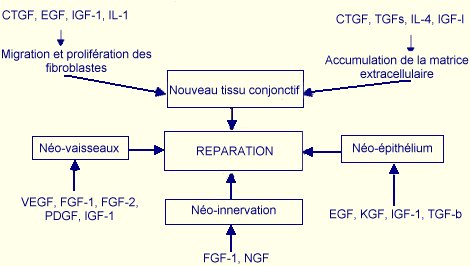
Figure 10Angiogénčse
Le remodelage tissulaire
Différentes matrices extracellulaires provisoires
Caillot de fibrine
Tissu de granulation
Matrice extracellulaire mature
Contraction du tissu cicatriciel
Protéinases et remodelage tissulaire
Généralités
Les métalloprotéinases matricielles (MMPs)
Classification
Structure
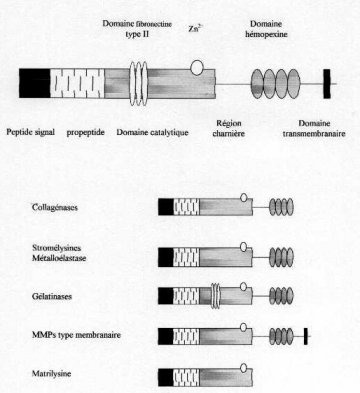
Figure 11Activation des MMPs
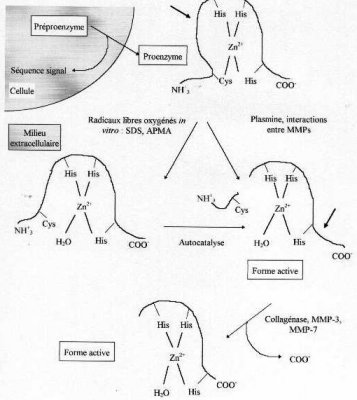
Figure 12Régulation de l’activité des MMPs
Régulation de l’expression
Régulation post-traductionnelle : les inhibiteurs des MMPs (TIMPs)
Rôles des collagénases
Rôles des stromélysines
Rôles des gélatinases
Le complexe Glycyl-L-Histidyl-L-Lysine-Cu2+ (GHK-Cu)
Structure
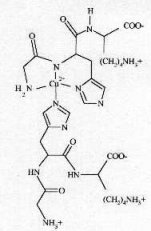
Figure 13Métabolisme, Distribution
Chimiotactisme
Facteur de croissance
Facteur angiogénique
Activité SOD-like
Synthčse de composants matriciels
GHK-Cu et cicatrisation
Etudes in vivo chez l’animal
Etudes cliniques chez l'homme
Développement industriel du GHK-Cu